The European Union (EU) is an economic and political union of 28 member states in Europe. It was founded in 1993 with the aim of creating a closer union between the peoples of Europe and has 23 official languages. The CE marking is mandatory for all products sold within the European Union that fall within the scope of CE marking directives.
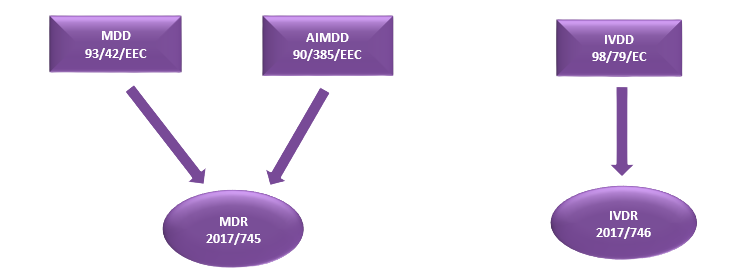
There are three main directives for medical devices:
Understanding the product and identifying applicable standards as per the intended purpose of the product.
Identifying applicable regulatory requirements and preparation of regulatory assessment report.
Compiling the technical documentation, product registration, and launch.
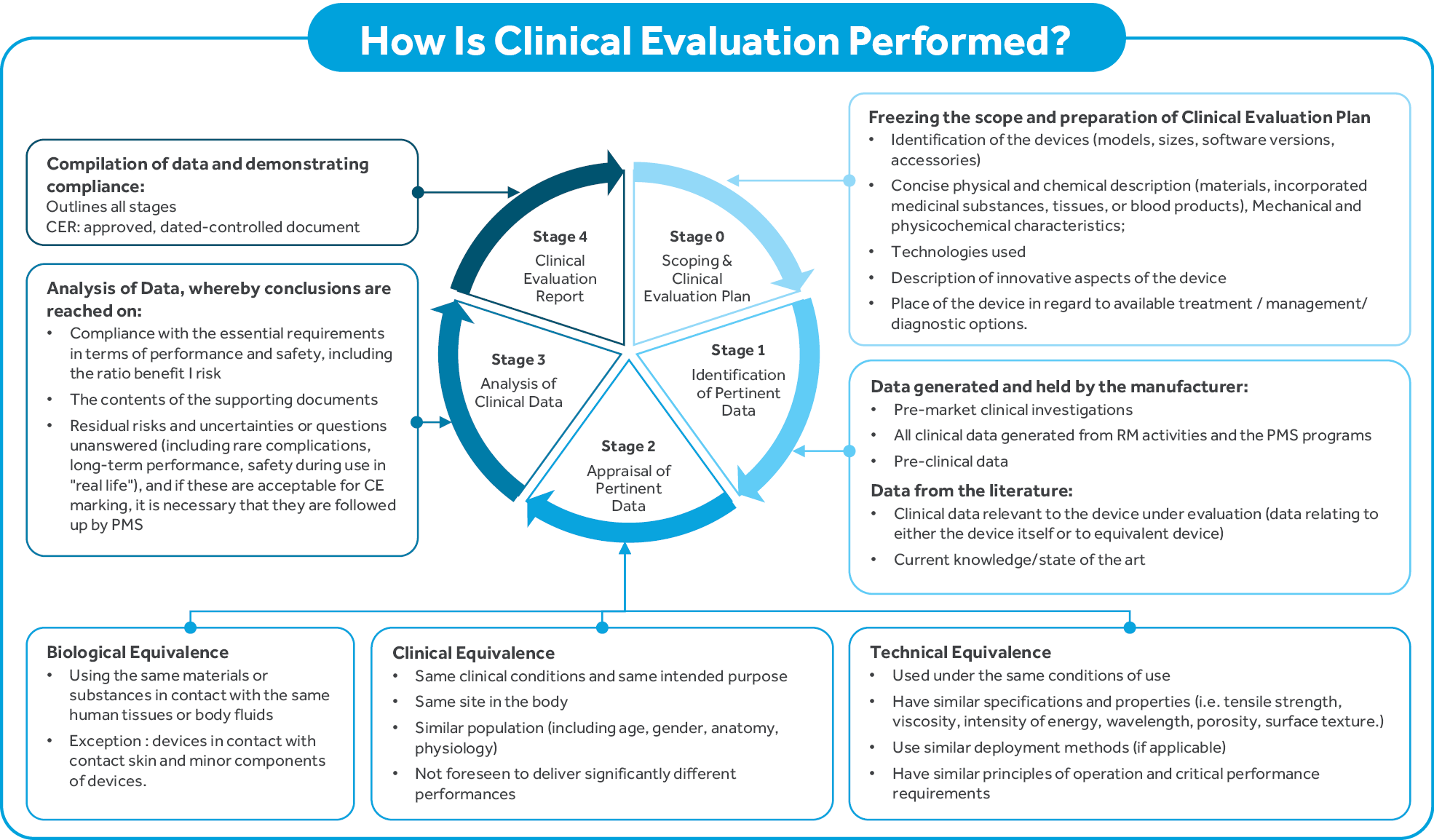
The clinical evaluation should be conducted by a suitably qualified individual or a team, taking into consideration:


A cloud-based solution to accelerate the regulatory compliance process. It helps to search worldwide regulations, and offers a digitized form regulation database for easy search and analysis. The standard module consists of a library of 1500+ international standards such as ISO/IEC/AAMI.
The solution offers a device classification tool, device-specific compliance, and regulatory intelligence services such as regulation assessment, gap assessment, and impact analysis.
Its regulatory watch feature monitors changes in regulations and provides a personalized news feed to users consisting of regulations news, safety communication, and warning letters.

Most medical device companies are racing against time to ensure compliance to EU MDR-217/745 where clinical evaluation is critical. Cyient offers a one-stop solution for helping medical device companies with clinical evaluation for regulatory compliance. Empowered by our Quality Assurance and Regulatory Affairs (QARA) CoE, Cyient has certified professionals across all functions who have the required skill sets and expertise to support medical device companies throughout the clinical evaluation of medical devices.

Abhishek Kumar is an SME in medical device regulatory and quality assurance services. With 12+ years of experience, he has successfully led multiple engagement programs for Europe, US, China, ASEAN markets for NPD and sustenance. Additionally, Abhishek has prepared and implemented the regulatory plan for NPD for 90+ countries by analyzing project feasibility, freezing regulatory requirements, and coordinating with various cross-functional teams.