Regulatory affairs play a vital role throughout the lifecycle of a medical device. It enables medical device manufacturers to devise premarket strategy, drafting regulatory submissions, and ensuring post-market compliance. Here is a quick look at the medical device regulatory affairs market share.

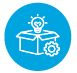
As per the current market trend, new product development (NPD), sustenance, and transition are the three main activities where a regulatory affairs specialist has a major role to play. As an example, let us consider the European market with regard to:
Understanding the product and identifying applicable standards as per the intended purpose of the product.
Identifying applicable regulatory requirements and preparation of regulatory assessment report.
Compiling the technical documentation, product registration, and launch.
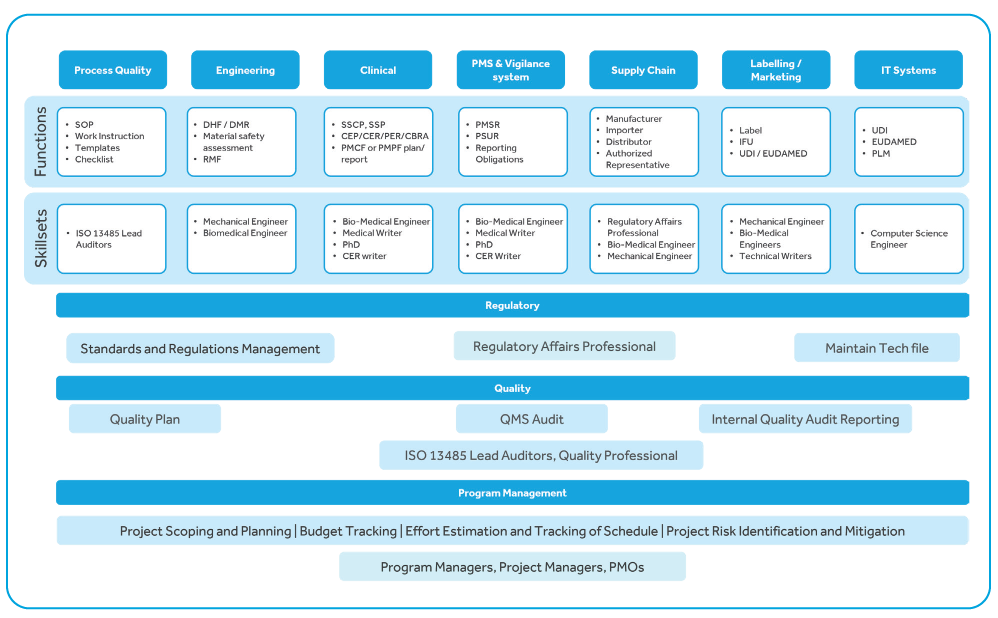
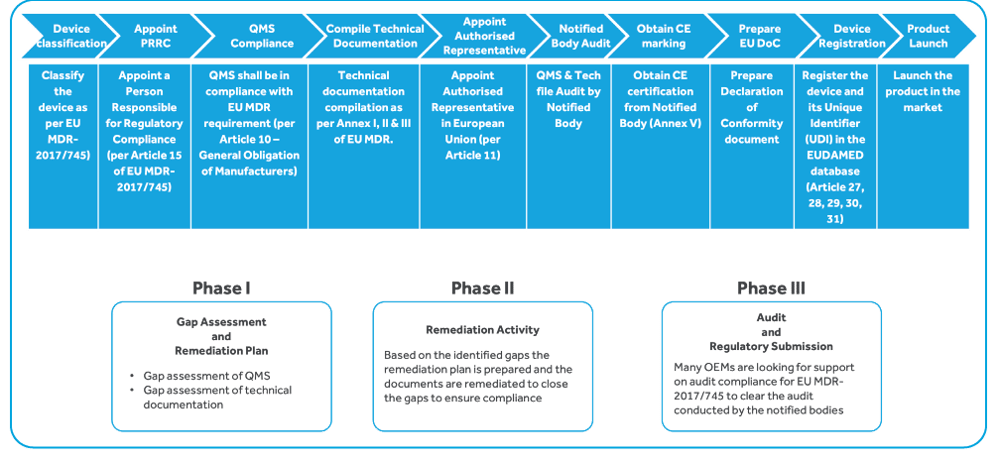
To continue selling medical devices in the European Union post MDR go-live, manufacturers have to perform
These regulations apply in the case of:
The following are covered by these regulations:

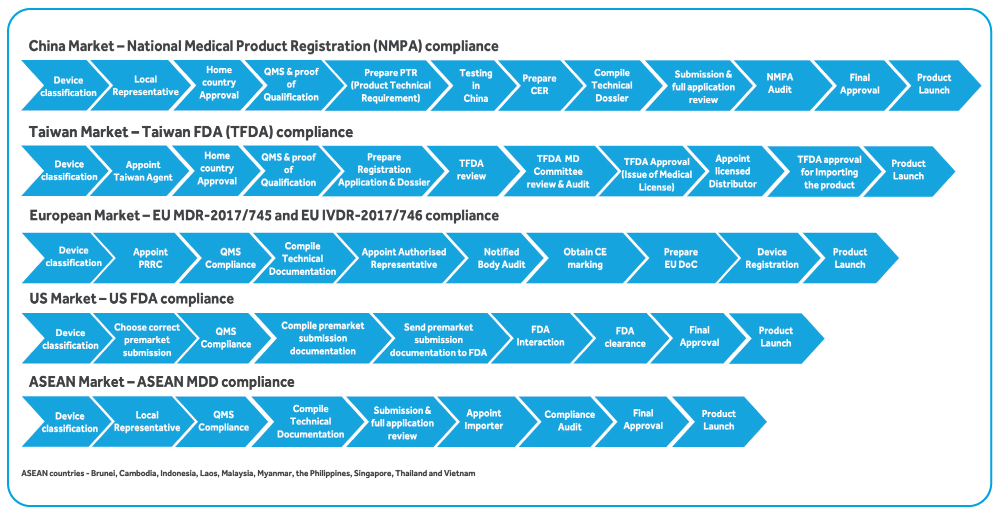
The diagram below represents Cyient’s understanding and function-wise approach for NPD, transition and sustenance. The thought-process behind the below representation is to clearly identify the requirements as per the applicable regulatory requirement and function-wise segregation of tasks/ activities to assign ownership to ensure compliance. The ownership is assigned to associates with right skillsets listed in the bucket below.

A cloud-based solution to accelerate the regulatory compliance process. It helps to search worldwide regulations, and offers a digitized form regulation database for easy search and analysis. The standard module consists of a library of 1500+ international standards such as ISO/IEC/AAMI.
The solution offers a device classification tool, device-specific compliance, and regulatory intelligence services such as regulation assessment, gap assessment, and impact analysis.
Its regulatory watch feature monitors changes in regulations and provides a personalized news feed to users consisting of regulations news, safety communication, and warning letters.

Abhishek Kumar is an SME in medical device regulatory and quality assurance services. With 12+ years of experience, he has successfully led multiple engagement programs for Europe, US, China, ASEAN markets for NPD and sustenance. Additionally, Abhishek has prepared and implemented the regulatory plan for NPD for 90+ countries by analyzing project feasibility, freezing regulatory requirements, and coordinating with various cross-functional teams.