Intelligent Engineering
Our Intelligent Engineering solutions across products, plant and networks, combine our engineering expertise with advanced technologies to enable digital engineering & operations, develop autonomous products & platforms, and build sustainable energy and infrastructure
.png?width=774&height=812&name=Master%20final%201%20(1).png)

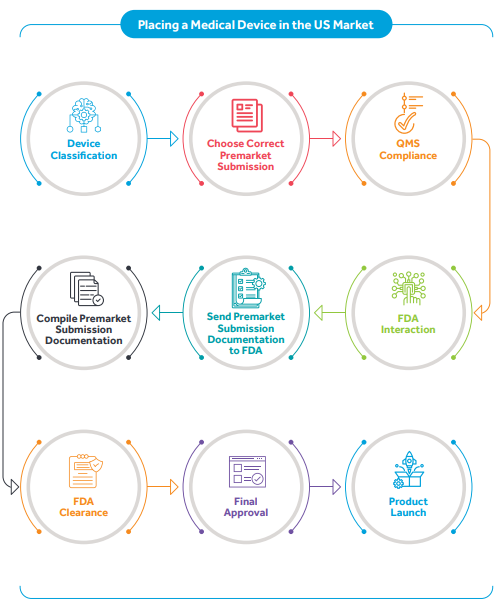
Introduction to USFDA Regulations
The United States Food and Drug Administration (FDA) is a regulatory agency responsible for protecting and promoting public health through the regulation and supervision of food safety, pharmaceuticals, medical devices, cosmetics, and other products. Established in 1906, the FDA ensures that these products are safe, effective, and properly labeled.
FDA regulations encompass a wide range of activities, including pre-market approval of new products, ongoing surveillance of marketed products, enforcement of manufacturing standards, and oversight of advertising and labeling.
For pharmaceuticals and medical devices, the FDA evaluates safety and efficacy through a rigorous review process before allowing them to be marketed. The agency also monitors post-market safety through adverse-event reporting and inspections of manufacturing facilities.
In the food industry, the FDA sets standards for food safety, labeling, and nutritional content to protect consumers from adulteration and misbranding. Additionally, the FDA regulates additives, pesticides, and contaminants in food products.
Overall, FDA regulations play a crucial role in safeguarding public health by ensuring the safety, efficacy, and quality of products available to consumers in the United States using the Code of Federal Regulations (CFR).

The Code of Federal Regulations
CFR, or the Code of Federal Regulations, is a compilation of all the rules and regulations promulgated by federal agencies in the United States. These regulations are organized into 50 titles, each covering different areas of governance, such as agriculture, commerce, transportation, and health.
The CFR is divided into chapters, parts, sections, and subsections, providing a detailed framework for implementing federal laws. It serves as a primary source of regulatory information for businesses, government officials, attorneys, and the general public.
Federal agencies use the CFR to publish regulations that have the force of law. These regulations undergo a process of notice and comment, allowing stakeholders to provide feedback before they are finalized. Once published, CFR regulations are enforceable by law enforcement agencies and carry penalties for non-compliance.
Overall, the CFR plays a crucial role in shaping and governing various aspects of public policy and regulatory compliance in the United States.

Overview of De Novo Submission
The De Novo submission provides a marketing pathway to classify novel medical devices for which general controls alone, or general and special controls, provide reasonable assurance of safety and effectiveness for the intended use, but for which there is no legally marketed predicate device.
De Novo classification is a risk-based classification process.
Devices that are classified into Class I or Class II through a De Novo classification request (De Novo request) may be marketed and used as predicates for future premarket notification [510(k)] submissions, when applicable.
When to prepare a De Novo submission request
There are two scenarios which prompt the submission of a De Novo request to the FDA to make a risk-based evaluation for classification of a device as Class I or Class II.
- Scenario - 1: Upon receiving a high-level not substantially equivalent (NSE) determination (that is, no predicate, new intended use, or different technological characteristics that raise different questions of safety and effectiveness) in response to a 510(k) submission.
- Scenario - 2: Upon the requester's determination that there is no legally marketed device upon which to base a determination of substantial equivalence (therefore without first submitting a 510(k) and receiving a high-level NSE determination).
Note: De Novo submission is not applicable for Class III devices

Key steps in the De Novo submission process
- Determining eligibility: Determine if the device is eligible for the De Novo pathway. Devices that are not Class III and do not have a predicate device are typically eligible.
- Preparation of submission: Prepare a comprehensive submission package, including a cover letter, device description, intended use, indications for use, device labeling, risk assessment, and proposed classification.
- Submit the De Novo request: Submit the De Novo request to the USFDA. This submission should include all required information and supporting documentation. The submission can be made electronically through the FDA's electronic submission gateway or by mailing a paper submission.
- FDA review: The FDA will review the De Novo request to determine whether your device meets the criteria for granting marketing authorization. This process includes an assessment of the device's safety and effectiveness, as well as its intended use and potential risks.
- Additional information requests: The FDA may request additional information or clarification during the review process. It's essential to respond promptly and thoroughly to any requests to expedite the review process.
- Decision: The FDA will decide on your De Novo request based on the review findings. If the request is granted, your device will be classified into either Class I or Class II, and you will receive marketing authorization to market the device in the United States.
- Post-market requirements: After receiving marketing authorization, you may be subject to post-market requirements, such as post-market surveillance, labeling requirements, and compliance with any special controls imposed by the FDA.

Content of a De Novo Submission
A De Novo request includes all the content elements necessary for acceptance of the De Novo request, listed in Appendix A of the "Acceptance Review for De Novo Classification Request" guidance document. The FDA intends to Refuse to Accept a De Novo request that does not include the following elements:
- Coversheet clearly identifying the request as a "Request for Evaluation of Automatic Class III Designation" under 513(f)(2) De Novo request.
- Administrative information, such as the device's intended use, prescription use or over-the-counter use designated, etc.
- Device description, which includes but is not limited to technology, proposed conditions of use, accessories, and components.
- Classification information and supporting data:
- Classification as per Section 513 of the Federal Food, Drug, and Cosmetic Act (FD&C Act) along with complete discussion of -
- Why general controls or general and special controls provide reasonable assurance of the safety and effectiveness of the device; and
- What special controls, if proposing a Class II designation, would allow the agency to conclude there is reasonable assurance the device is safe and effective for its intended use.
- Clinical data (if applicable) that are relevant to support reasonable assurance of the safety and effectiveness of the device.
- Non-clinical data including bench performance testing.
- Information on –
- Reprocessing and sterilization
- Shelf life
- Biocompatibility and material information
- Software
- Electrical safety and electromagnetic compatibility
- Animal study literature (if applicable)
- Risk assessment, which includes description of the probable benefits of the device when compared to the probable or anticipated risks when the device is used as intended.
- Labeling and instructions for use.
- Quality management system (QMS) documentation demonstrating compliance with 21 CFR Part 820 and applicable international quality standards.
- Declaration of conformity.

Considerations for successful De Novo submission
Early engagement with FDA: It is advisable to open communication with the FDA early in the development process which can help clarify regulatory expectations, identify potential challenges, and streamline the submission process by taking the Pre-Submission (Pre-Sub) route.
Comprehensive data generation: Generating robust and scientifically sound data to support the device's safety and effectiveness is crucial for a successful De Novo submission. Investing in clinical studies, when appropriate, can strengthen the submission and enhance the likelihood of FDA approval.
Regulatory strategy: Developing a comprehensive regulatory strategy that aligns with the device's intended use, target market, and commercialization timeline is essential for navigating the regulatory pathway effectively and efficiently.
Post-market considerations: Planning for post-market obligations, including post-market surveillance, quality management, and labeling updates, is essential for maintaining compliance and ensuring ongoing patient safety and device effectiveness.

Pre-Submission and Its Usefulness
A Pre-Submission or Pre-Sub is a type of Q-Submission (Q-Sub) which is a formal written request from a submitter for feedback from FDA that is provided in the form of a formal written response or, if the submitter chooses, formal written feedback followed by a meeting. All discussion that occurs during the meeting is documented in meeting minutes that are drafted by the submitter and submitted for FDA review.
A Pre-Sub provides the opportunity to obtain FDA feedback prior to an intended premarket submission—Investigational Device Exemption (IDE), Premarket Approval (PMA), Humanitarian Device Exemption (HDE), De Novo request, 510(k), Dual 510(k) and CLIA Waiver by Application Submissions (Duals), Biologics License Applications (BLAs), Investigational New Drug Applications (INDs)), Accessory Classification Request, or Waiver by Applications (CW).
The program is entirely voluntary on the part of the submitter. However, early interaction with FDA on planned non-clinical and clinical studies and careful consideration of FDA’s feedback may improve the quality of subsequent submissions, shorten total review times, and facilitate the development process for new devices. FDA believes that interactions provided within Pre-Subs are likely to contribute to a more efficient and transparent review process for FDA and the submitter. FDA develops feedback for Pre-Subs by considering multiple scientific and regulatory approaches consistent with least burdensome requirements and principles, to streamline regulatory processes. FDA has found that feedback is most effective when requested prior to execution of planned testing. Issues raised by FDA in a Pre-Sub do not obligate submitters to address or resolve those in a subsequent submission, though any future submission related to that topic should discuss why a different approach was chosen or an issue left unresolved. Further, review of information in a Pre-Sub does not guarantee a favorable decision in future submissions. Additional questions may be raised during the review of the future submission when all information is considered as a whole, or if new information has become available since the Pre-Sub.

Types of Q-Submission

FDA’s Final Actions on a De Novo Request
After review of the De Novo request, the FDA will make a final decision to either grant or decline. FDA will also consider the De Novo request to be withdrawn in certain situations.
-
Grant De Novo Request (21 CFR 860.260)
If the data and information provided to the FDA demonstrate that general controls or general and special controls are adequate to provide reasonable assurance of safety and effectiveness, and the probable benefits of the device outweigh the probable risks, then the FDA may grant the De Novo request and establish a new classification regulation for the new device type.
-
Decline De Novo Request (21 CFR 860.260)
De Novo request is declined by the FDA if –
- General controls or general and special controls are insufficient to provide reasonable assurance of safety and effectiveness of the device
- The data provided in the De Novo request are insufficient to determine whether general controls or general and special controls can provide a reasonable assurance of safety and effectiveness of the device
- The probable benefits of the device do not outweigh the probable risks
If the De Novo request is declined, the device remains in Class III and the requester may not legally market the device. The FDA will issue a written order to the requester identifying the reasons, which can include lack of performance data that warrant declining the De Novo request. The requester should generally either submit an application for premarket approval under Section 515 of the FD&C Act or collect additional information to address the issues and submit a new De Novo request that includes the additional information.
-
Withdrawal of a De Novo Request (21 CFR 860.250)
The FDA will consider a De Novo classification request to be withdrawn if—
- The requester submits a written notice to the FDA that the requester is withdrawing the De Novo request;
- The requester fails to provide a complete response to a request for additional information (21 CFR 860.240), or deficiencies identified by the FDA (21 CFR 860.230) are not addressed within 180 days after the date the FDA issues such request; or
- The requester does not permit an authorized FDA employee an opportunity to inspect the facilities (21 CFR 860.240), at a reasonable time and in a reasonable manner, and to have access to copy and verify all records pertinent to the De Novo request.
If the FDA considers a De Novo request to be withdrawn, the FDA notifies the requester with reference to the De Novo request number and the date the FDA considered the De Novo request withdrawn.

FDA's Review Process and Timeline for De Novo Submission Request
- Acceptance review (21 CFR 860.230)
Upon receipt of a De Novo request, the FDA will conduct an acceptance review. The acceptance review is an administrative review to assess the completeness of the application and whether it meets the minimum threshold of acceptability. If any of the acceptance elements are not included, a justification has to be provided for the omission.
To aid in the acceptance review, it is recommended to submit an Acceptance Checklist as per the guidance document with the De Novo request that identifies the location of supporting information for each checklist element.
The De Novo request will not be accepted and will receive a Refuse to Accept (RTA) designation if one or more of the elements noted as RTA items in the Acceptance Checklist are not present and no explanation is provided for the omission(s). However, during the RTA review, the FDA staff has the discretion to determine whether the missing checklist elements are needed to ensure the De Novo request is administratively complete to allow the De Novo request to be accepted.
Within 15 calendar days of the Document Control Center receiving the De Novo request, the FDA will notify the requester electronically of the acceptance review result as one of the following:
- The De Novo request has been accepted for substantive review;
- The De Novo request has not been accepted for review (i.e., considered RTA) and the requester has 180 calendar days to fully address the RTA notification; or
- The De Novo request is under substantive review and the FDA did not complete the acceptance review within 15 calendar days.

- Substantive review
Once the De Novo request is accepted for substantive review, the FDA conducts a classification review of legally marketed device types and analyzes whether an existing legally marketed device of the same type exists. This information is used to confirm that the device is eligible for De Novo classification.
During the substantive review of a De Novo request, the FDA may identify deficiencies that can be adequately addressed through interactive review and not require a formal request for additional information.
If the issues and deficiencies cannot be addressed through interactive review, an Additional Information letter will be sent to the requester. If an Additional Information letter is sent, then the De Novo request will be placed on hold. The requester has 180 calendar days from the date of the Additional Information letter to submit a complete response to each item in the Additional Information letter.
Note: The response must be sent to the DCC within 180 calendar days of the date of the Additional Information letter. No extensions beyond 180 days are granted. If the FDA does not receive a complete response to all deficiencies in the Additional Information letter within 180 days of the date of the letter, the request will be considered withdrawn and deleted from the FDA's review system. If the De Novo request is deleted, the De Novo requester will need to submit a new request to pursue the FDA's marketing authorization for that device.
The requester must submit their response to an Additional Information letter in electronic format (eCopy), to the DCC of the appropriate center. The response should—
- Include the requester's name;
- Identify the De Novo number;
- Include the requester's name;
- Identify the submission as a response to the Additional Information letter;
- Identify the date of the FDA's request for additional information; and
- Provide the requested information in an organized manner.
The final step is the De Novo request decision. Under MDUFA IV, the FDA's goal is to decide about a De Novo request in 150 review days. Review days are calculated as the number of calendar days between the date the De Novo request was received by the FDA and the date of the FDA's decision, excluding the days a request was on hold for an Additional Information request.

CyARC—Accelerated Regulatory Platform
Cyient offers a one-stop solution, CyARC–Accelerated Regulatory Platform, for helping medical device companies to ensure regulatory compliance. Empowered by Quality Assurance and Regulatory Affairs (QARA) CoE, Cyient has certified professionals across all the functions who have the required skillsets and expertise to support medical device companies throughout the life-cycle of their medical devices.

About the Author

Abhishek Kumar is an SME in Medical Device Regulatory Affairs, Quality Assurance, and Clinical Affairs with over 13 years of experience. He has led the EU MDR-2017/745 sustenance program, identifying business opportunities for sales teams, and managed the engagement program for a US-based medical device company. He has supported the gap assessment, remediation, and submission of 45+ Technical Documentations as per EU MDR, and created 40+ CERs for Class I, II, and III medical devices according to MEDDEV 2.7.1 Rev-4. Additionally, Abhishek has developed proposals for global markets, including Europe, US, ASEAN, China, Taiwan, and Japan, and prepared and implemented regulatory plans for NPD in 90+ countries by analyzing feasibility, defining requirements, and coordinating cross-functional teams.
