
- 2026-Jun-04
Espen Berg
Our Intelligent Engineering solutions across products, plant and networks, combine our engineering expertise with advanced technologies to enable digital engineering & operations, develop autonomous products & platforms, and build sustainable energy and infrastructure
.png?width=774&height=812&name=Master%20final%201%20(1).png)
This is Part 1 of 7-part series explaining the different scenarios and use cases of Human Factors in Medical Devices.
Medical devices were once as large as an entire room. However, devices today are smaller than a human palm and have seen a significant shift in form factor. While medical devices are transitioning into modern, user-friendly forms such as smartwatches, compact devices, home-based tests, handheld scanners, and implants, these transformations are also introducing new USE challenges for users. These challenges often lead to USE errors due to consistent changes that these technologies undergo.
Consider these real-life incidents that shed light on the importance of addressing user-related concerns in medical devices:
Many medical devices are developed without incorporating Usability Engineering (Human Factors Engineering) Process. As a result, these devices become non-intuitive, challenging to learn, and difficult to use. As healthcare practices evolve, people with the most limited medical expertise, including patients themselves, are using these devices, making usability engineering increasingly crucial.
In an effort to tackle safety complaints related to device usage, developed countries like the United States and the European Union are placing significant emphasis on designing medical devices using usability engineering processes. This approach involves establishing integration points with product realization and risk management procedures. It's important to note that user-friendly design is not simply a matter of artistic flair but rather the result of a systematic engineering process.
The usability engineering process comprises key elements that collectively ensure the development of user-friendly medical devices. These elements include:

Regulatory bodies, such as the US FDA and the European Union's Medical Device Regulation (EU MDR), have recognized the importance of the IEC 62366-1:2015 standard and its subsequent amendments. Manufacturers of medical devices are now required to document the history of usability design in a Usability Engineering File, which plays a pivotal role in regulatory submissions, including CE certification and 510(k) approvals.
The Usability Engineering File comprises several essential components, each of which plays a crucial role in ensuring the usability and safety of medical devices. These components include:
In this series, we will discuss each of these components in a separate article.
Understanding Human Factors
In the context of medical device usability, human factors, or usability engineering, focuses on the interactions between individuals and the devices they use.
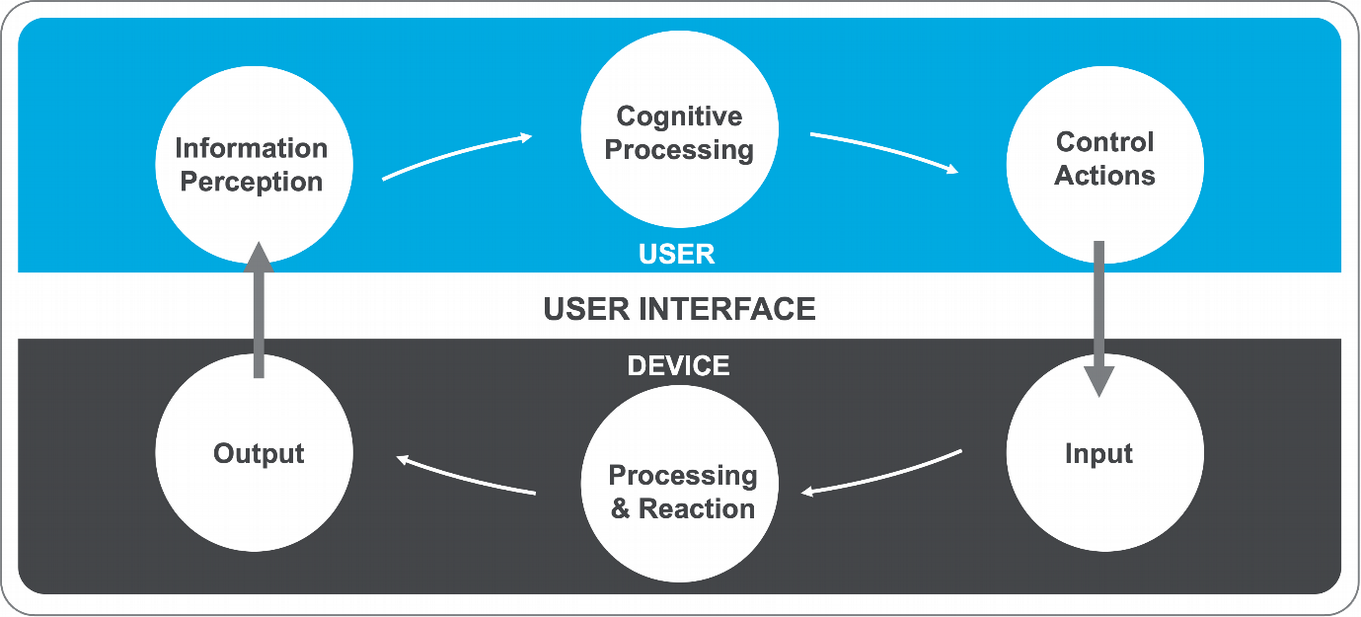
This interaction encompasses how users:
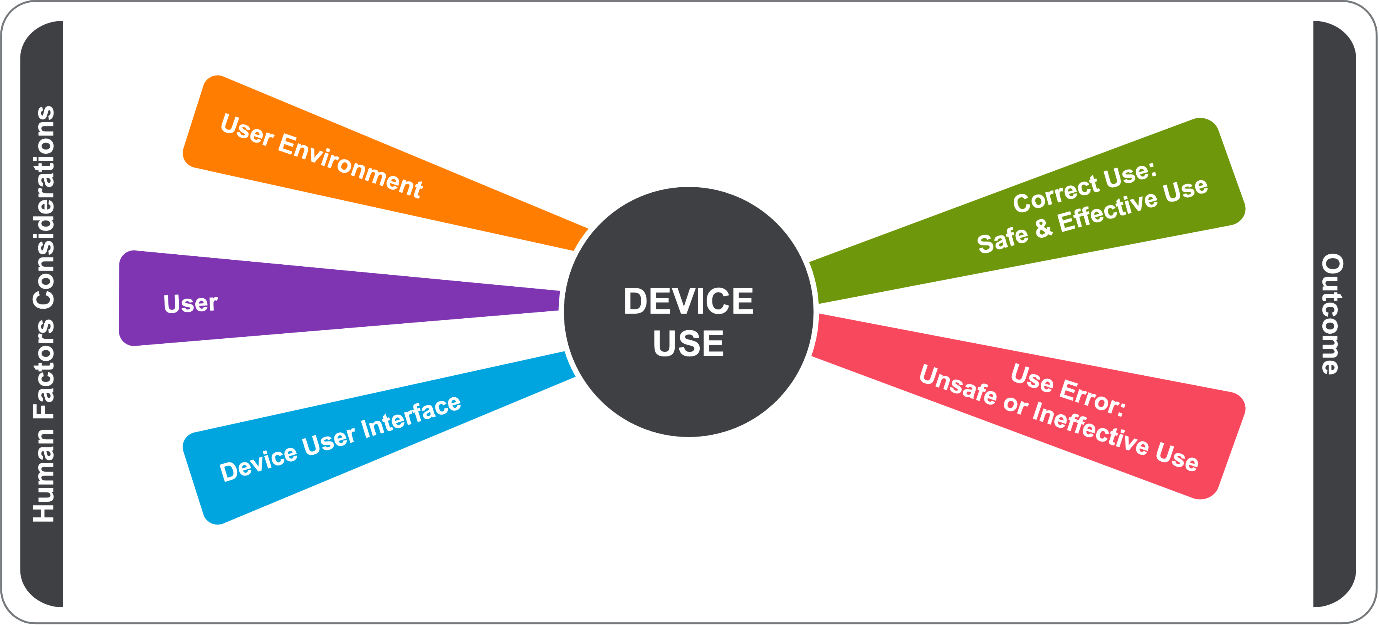
Human factors/usability engineering is used to design the user-device interface. The user interface includes all components with which users interact while preparing the device for use (e.g., unpacking, set up, calibration), using the device, or performing maintenance (e.g., cleaning, replacing a battery, making repairs).
According to the FDA, the primary objective of human factors/usability engineering in medical devices is to minimize use-related hazards and risks. This ensures that users can safely and effectively use the device. Benefits of applying human factors principles include:
Intuitive and error-free design requires intensive user research and references to standards, guidelines and rules of thumb. Let us explore the US FDA regulation, guidance documents and standards available in this context.
The US FDA has incorporated human factors-related requirements into its Quality System Regulations (21 CFR Part §820.30 Design Controls). This integration encompasses various aspects, including user needs assessment, usability verification, and validation. The FDA also provides guidance documents to assist manufacturers in implementing human factors and usability engineering processes effectively.
In addition to the core regulations and standards mentioned above, there are several other important regulatory documents that impact the development and evaluation of medical devices:
Designing medical devices with human factors considerations is fundamental to ensuring their safety, usability, and effectiveness. In upcoming articles, we will delve deeper into the usability procedure, including the analysis of use environments, definition of use scenarios, identification of frequently used functions, and derivation of user interface requirements, as outlined in the IEC 62366-1:2015 standard.
In the next part of this series, we discuss how to analyze USE environment, define USE scenarios, frequently USED functions and derive user interface requirements. We will also explore the design input document - USER INTERFACE SPECIFICATION as explained in IEC 62366-1:2015 standard.
About the Author
Aboli is a Solution Architect, working for Healthcare and Life sciences, CTO Group. An articulate and experienced professional with 18+ years of demonstrative experience in requirements engineering, usability engineering, product risk management, software and hardware testing, verification and validation in Life Sciences and Medical Device domains. Played crucial role in the research, design, development and release of complex Medical Device systems and Clinical Trial related IT applications to deliver secure, compliant, and efficient solutions. Currently, building a solution accelerator "CyARC" with an objective to automate medical device regulatory compliance processes powered by artificial intelligence.



